Marius Pachitariu and Carsen Stringer released a Python version of their Suite2p pipeline for two-photon imaging. So I decided to take this tool for a test drive with data collected from Scanbox.
At the moment, Suite2p takes tiff and hdf5 files, so we first decided to write a function to convert *.sbx to *.h5 files. The sbx2h5 function below, does just that:
function sbx2h5(fname,varargin)
% sbx2h5
% Generates h5 file from sbx files
fnh = [fname ,'.h5'];
z = sbxread(fname,1,1);
global info;
if(nargin>1)
N = min(varargin{1},info.max_idx);
else
N = info.max_idx;
end
k = 0;
done = 0;
blksize = 200; % block size
to_read = min(blksize,N-k);
while(~done && to_read>0)
try
q = sbxread(fname,k,to_read);
q = squeeze(q(1,:,:,:)); % extract green channel only
q = permute(q,[2 1 3]);
if(k==0)
h5create(fnh,'/data',[796 512 Inf],'DataType','uint16','ChunkSize',[796 512 to_read]);
h5write(fnh,'/data',q,[1 1 1],[796 512 to_read]);
f = waitbar(0,'Converting to hdf5');
else
h5write(fnh,'/data',q,[1 1 k+1],[796 512 to_read]);
end
catch
done = 1;
delete(f);
end
k = k+to_read;
to_read = min(blksize,N-k);
waitbar(k/N,f);
end
delete(f);
For now the function extracts only the green channel (PMT0) and assumes frames with 512 lines. If you have a Scanbox binary file, such as xx0_000_000.sbx, simply call sbx2h5(‘xx0_000_000’) in Matlab and you will get back an ‘xx0_000_000.h5’ file you can load directly into Suite2p.
If you then run Suite2p you will end up with a GUI showing the segmented cells:
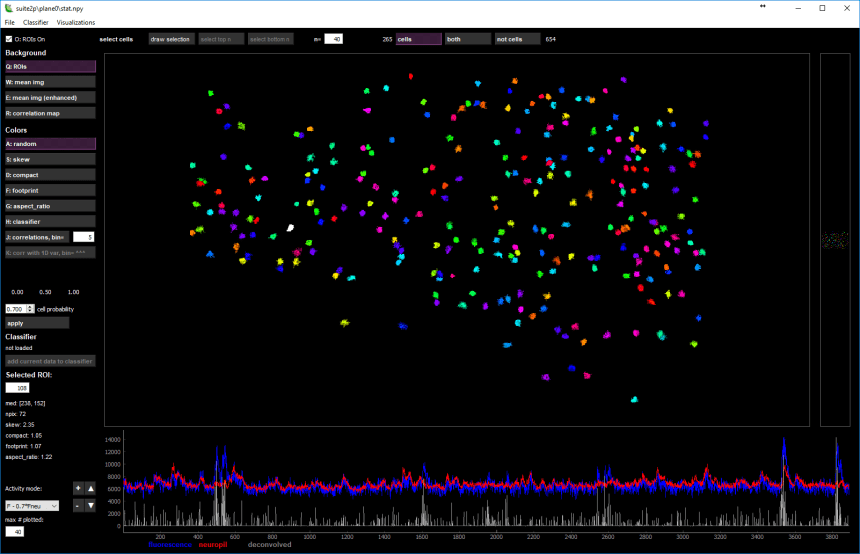
You also have to ask Suite2p to generate a Matlab output file (there is a field for that), and the results will be written into the file “Fall.mat”. Because nearly all Scanbox users already have scripts that take as input *.signals and *.segment files in their analyses, I wrote a function, sbxsuite2sbx that will take the Suite2p output and create these Scanbox files for you. This allows you to try Suite2p with the set of analysis functions you already develop for Scanbox files.
</pre>
function sbxsuite2sbx(smat,fn)
% Convert a suite2p segmentation to Scanbox format
sz = [512 796];
mask = zeros(sz);
s2p=load(smat);
k = 1;
spks = [];
sig = [];
np = [];
for i=1:length(s2p.stat)
if(s2p.iscell(i,1))
mask(sub2ind(sz,s2p.stat{i}.ypix,s2p.stat{i}.xpix)) = k;
spks(:,k) = s2p.spks(i,:)';
sig(:,k) = s2p.F(i,:);
np(:,k) = s2p.Fneu(i,:)';
k = k+1;
end
end
save([fn '.segment'],'mask');
save([fn '.signals'],'spks');
<pre>
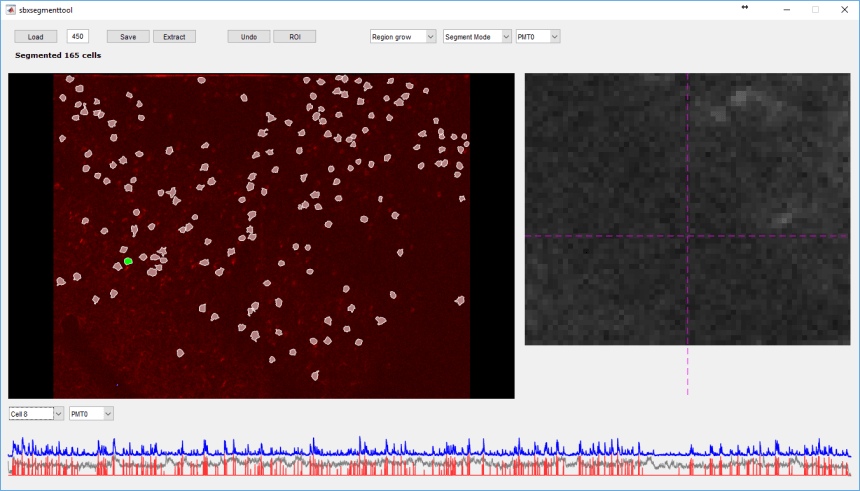
As an example, we segmented the same data as above using sbxaligntool and sbxsegmenttool in Scanbox:
We then matched the cells in the Scanbox and Suite2p segmentations using sbxmatchfield():

Here, yellow ROIs are the ones segmented by both methods; green outlines are ROIs segmented in Scanbox but not Suite2p; red outlines are ROIs segmented by Suite2p but not Scanbox (the outlines of nearby cells that overlap appear merged in this image).
Finally, we compared the analysis of our data using the spiking estimated with Suite 2p and with Scanbox on ROIs detected by both. This particular experiment involved the mapping of ON/OFF sub-regions in simple cells of mouse V1. Here are a few examples of such comparison:
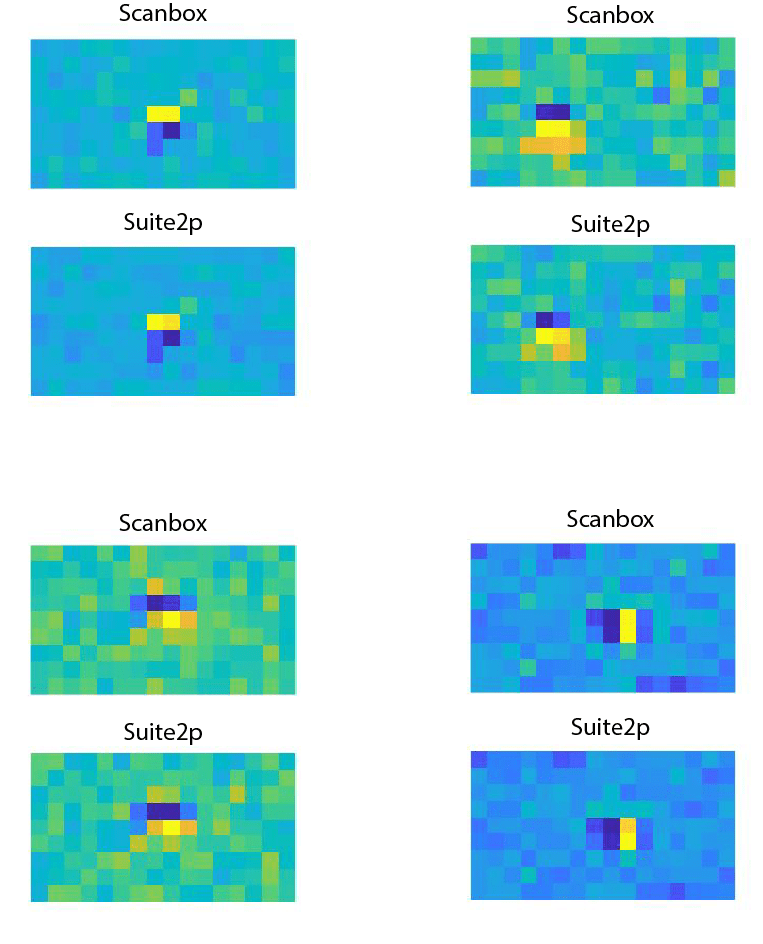
Each panel shows the receptive field of a simple cell estimated using the spikes from Scanbox or Suite2p. ON subregions appear in red, OFF sub-regions appear in blue. The results are nearly identical.
I have not checked the quality of the kernels for cells that Suite2p detects which were not segmented in Scanbox. At first sight, it seems Suite2p can detect more ROIs than using sbxsegmenttool, so it will certainly be worth checking and this could improve data yield. I will experiment a bit more and report about this soon.
The two functions above need a bit of work so they also work for arbitrary sizes, multiple ETL planes, and mesoscope ROIs. I will be including these in future releases of Scanbox but you can begin experimenting with Suite2p in the meantime.
If you have any questions about Suite2p, please direct them to Marius and Carsen.
If you have questions about using bridge functions above, post them in the comments below.
